Compare with chromVAR: Part I¶
Since we completely re-implemented the algorithm proposed in chromVAR in Python, we may want to confirm that these two packages can produce the similar, if not exactly same, results. So here we test them on a small example data from chromVAR.
Run chromVAR on example data¶
We first load the packages that we need
[1]:
suppressMessages(library(chromVAR))
suppressMessages(library(Repitools))
suppressMessages(library(SummarizedExperiment))
suppressMessages(library(motifmatchr))
suppressMessages(library(Matrix))
suppressMessages(library(BiocParallel))
suppressMessages(library(BSgenome.Hsapiens.UCSC.hg19))
suppressMessages(library(JASPAR2020))
suppressMessages(library(TFBSTools))
set.seed(2017)
Let’s get the example count matrix
[2]:
data(example_counts, package = "chromVAR")
We can check how the peaks look like:
[3]:
df_peaks <- annoGR2DF(example_counts@rowRanges)
df_peaks$peak <- paste0(df_peaks$chr, "-", df_peaks$start, "-", df_peaks$end)
head(df_peaks)
| chr | start | end | width | score | qval | name | peak | |
|---|---|---|---|---|---|---|---|---|
| <fct> | <int> | <int> | <int> | <int> | <dbl> | <chr> | <chr> | |
| 1 | chr1 | 713933 | 714432 | 500 | 259 | 25.99629 | GM_peak_6 | chr1-713933-714432 |
| 2 | chr1 | 856395 | 856894 | 500 | 82 | 8.21494 | H1_peak_7 | chr1-856395-856894 |
| 3 | chr1 | 895780 | 896279 | 500 | 156 | 15.65456 | GM_peak_17 | chr1-895780-896279 |
| 4 | chr1 | 911578 | 912077 | 500 | 82 | 8.21494 | H1_peak_13 | chr1-911578-912077 |
| 5 | chr1 | 948576 | 949075 | 500 | 140 | 14.03065 | GM_peak_27 | chr1-948576-949075 |
| 6 | chr1 | 954541 | 955040 | 500 | 189 | 18.99529 | GM_peak_29a | chr1-954541-955040 |
Next, we pre-process the data by adding GC bias for each peak and identify the backgrounds
[4]:
example_counts <- addGCBias(example_counts,
genome = BSgenome.Hsapiens.UCSC.hg19)
bg_peaks <- getBackgroundPeaks(example_counts)
write.csv(bg_peaks, "bg_peaks.csv")
Perform motif matching to get TF binding sites
[5]:
opts <- list()
opts[["collections"]] = c("CORE")
opts[["tax_group"]] = c("vertebrates")
motifs <- getMatrixSet(JASPAR2020, opts)
names(motifs) <- paste(names(motifs), name(motifs), sep = ".")
motif_ix <- matchMotifs(motifs, example_counts,
genome = BSgenome.Hsapiens.UCSC.hg19,
p.cutoff = 5e-05)
We here save the motif matching results for later use
[6]:
df_motifmatch <- as.data.frame(as.matrix(assays(motif_ix)$motifMatches) * 1)
write.csv(df_motifmatch, "chromvar_motif_match.csv")
We now can estimate the deviations with chromVAR
[7]:
dev <- computeDeviations(object = example_counts,
annotations = motif_ix)
dev
class: chromVARDeviations
dim: 746 50
metadata(0):
assays(2): deviations z
rownames(746): MA0004.1.Arnt MA0006.1.Ahr::Arnt ... MA0528.2.ZNF263
MA0609.2.CREM
rowData names(3): name fractionMatches fractionBackgroundOverlap
colnames(50): singles-GM12878-140905-1 singles-GM12878-140905-2 ...
singles-H1ESC-140820-24 singles-H1ESC-140820-25
colData names(2): Cell_Type depth
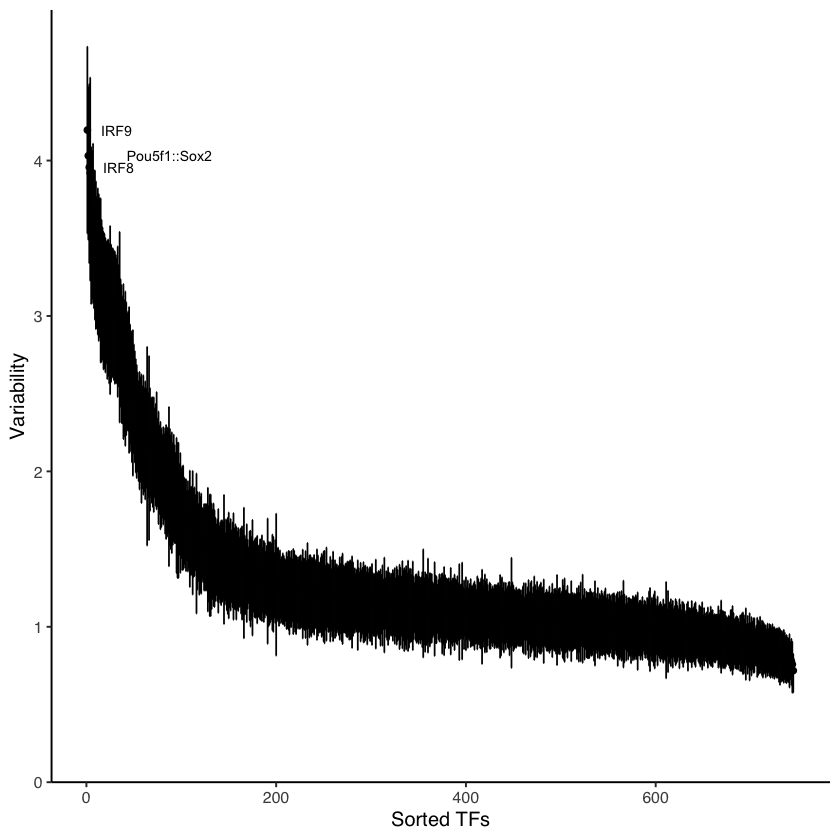
Plot variability
[10]:
variability <- computeVariability(dev)
plotVariability(variability, use_plotly = FALSE)
Warning message:
“Removed 1 rows containing missing values (`geom_point()`).”
[13]:
head(variability)
| name | variability | bootstrap_lower_bound | bootstrap_upper_bound | p_value | p_value_adj | |
|---|---|---|---|---|---|---|
| <chr> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | |
| MA0004.1.Arnt | Arnt | 1.3097587 | 1.0574367 | 1.539051 | 1.354635e-03 | 5.368102e-03 |
| MA0006.1.Ahr::Arnt | Ahr::Arnt | 1.6408099 | 1.3310370 | 1.897313 | 1.558287e-09 | 1.055386e-08 |
| MA0019.1.Ddit3::Cebpa | Ddit3::Cebpa | 0.9217392 | 0.7530152 | 1.071486 | 7.633490e-01 | 8.387992e-01 |
| MA0029.1.Mecom | Mecom | 1.1118984 | 0.9152195 | 1.279191 | 1.241110e-01 | 2.357689e-01 |
| MA0030.1.FOXF2 | FOXF2 | 1.2476069 | 0.9803963 | 1.488305 | 7.572098e-03 | 2.648457e-02 |
| MA0031.1.FOXD1 | FOXD1 | 1.4145492 | 1.1059477 | 1.689789 | 4.012849e-05 | 2.019982e-04 |
Let’s save the final results so we can compare it with pychromVAR
[8]:
df_z <- as.data.frame(t(round(assays(dev)$z, 2)))
write.csv(df_z, "chromvar_z.csv")
We also save the count matrix so we can use it as input for pychromVAR
[9]:
counts <- assays(example_counts)$counts
rownames(counts) <- df_peaks$peak
counts <- as.data.frame(as.matrix(counts))
write.csv(counts, "counts.csv")